ACM_BCB
Introduction to Epigenetic Clocks
What is Epigenetics?
Epigenetics refers to chemical modifications to DNA and its associated regulatory components that has the potential to influence gene transcription depending on genomic context, without altering the underlying DNA sequence.
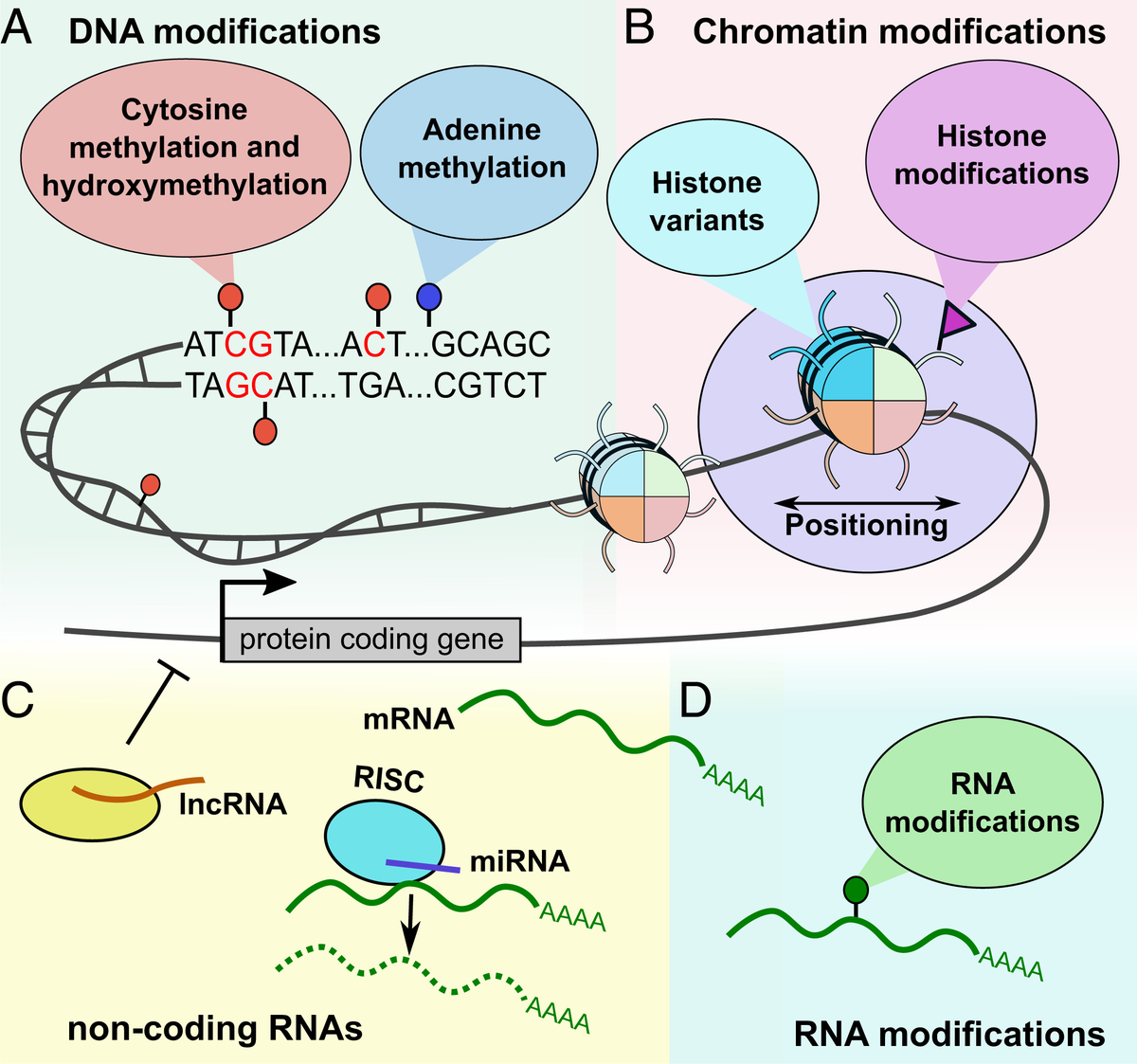
Figure 1. Overview of common epigenetic modifications (Aristizabal et al. (2019).
What is DNA Methylation?
DNA methylation is the addition of a methyl group to the 5’ carbon of the cytosine base, typically occurring at cytosine-phosphate-guanosine (CpG) dinucleotides. DNAm is the most widely studied epigenetic mark in human population and epidemiological research. —
What are the typical drivers of DNA Methylation Variation?
DNA methylation patterns vary substantially across individuals and are influenced by several biological factors including (not limited to):
- Tissue source
- Cell-type composition
- Genetic ancestry
- Sex
- Age
What are Epigenetic Clocks?
Epigenetic clocks are DNA methylation-based biomarkers that use machine learning algorithms to predict chronological age and/or biological age from DNA methylation data.
- Absolute Age Acceleration (AAA) or Epigenetic Age Difference (EAD): Difference between DNA methylation age and chronological age.
- Relative Age Acceleration (RAA) or Epigenetic Age Acceleration (EAA): Residuals obtained from regressing DNA methylation age on chronological age.
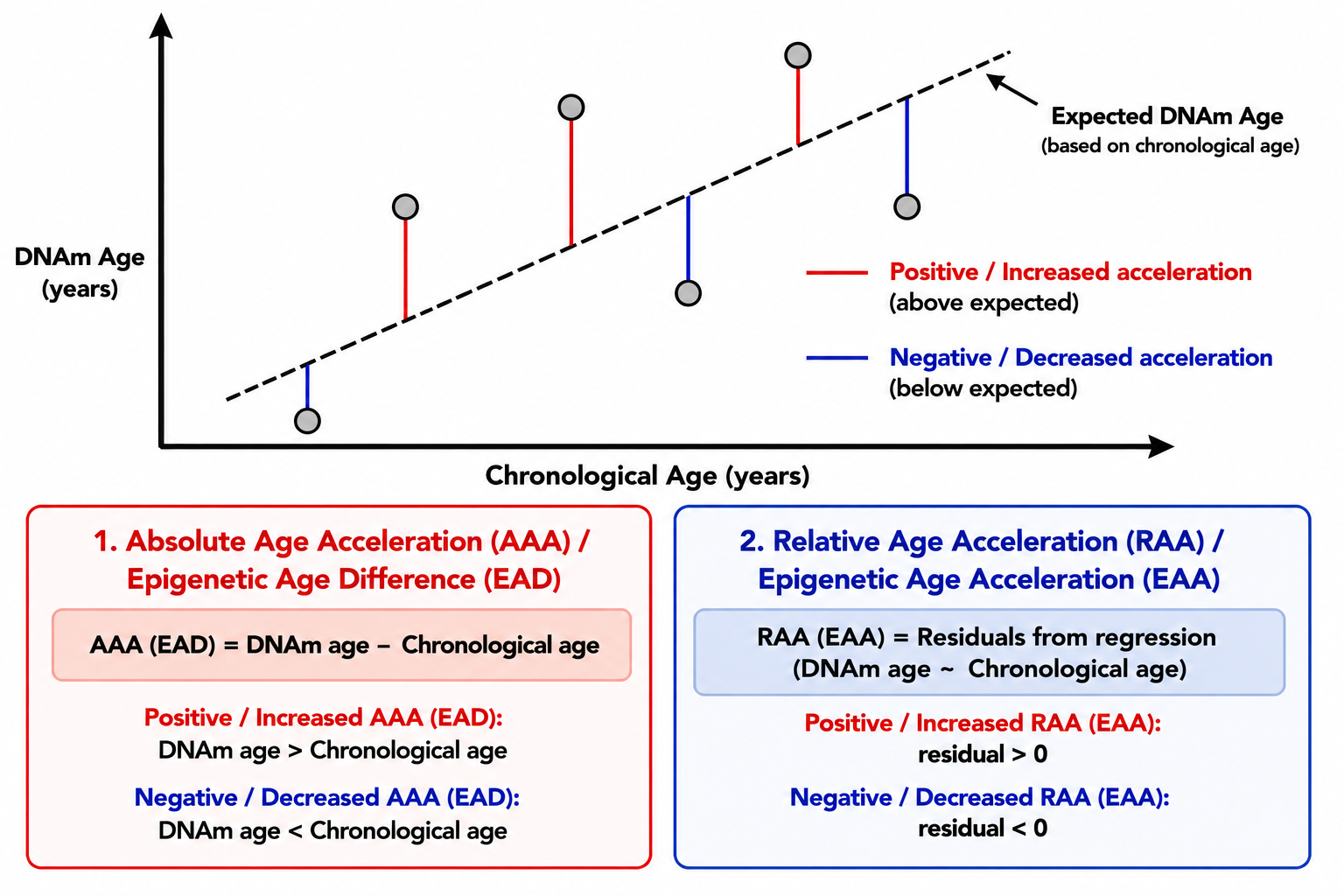
Figure 2. Epigenetic aging concepts.
First-Generation Clocks
Trained exclusively on chronological age.
Examples
- Horvath Pan-Tissue Clock
- Hannum Clock
Second-Generation Clocks
Trained on chronological age while incorporating age-related phenotypes associated with morbidity and mortality.
Examples
- PhenoAge
- GrimAge
Third-Generation Clocks
Developed using longitudinal cohorts and trained to estimate the pace of biological aging rather than chronological age itself.
Example
- DunedinPACE
Factors Influencing Epigenetic Aging
Epigenetic age estimates can be influenced by several biological including tissue source, cell-type composition, sex, and genetic ancestry.
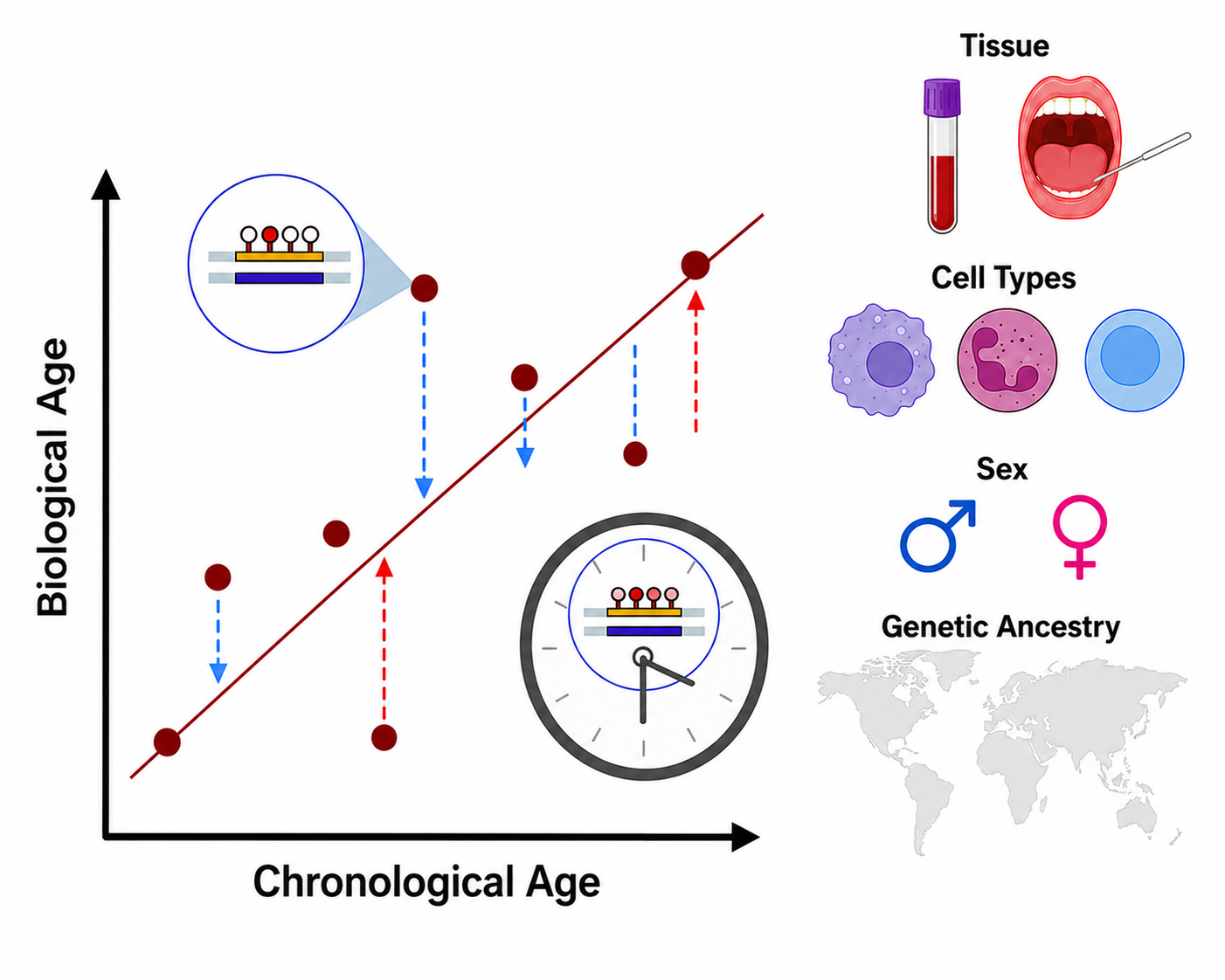
Figure 3. Biological and technical factors that influence epigenetic age estimates (Adapted and modified from materials developed by the Merrill Lab).
Epigenetic Age Difference and Epigenetic Age Acceleration
The discrepancy between DNA methylation-predicted age and chronological age is commonly used as a measure of biological aging.
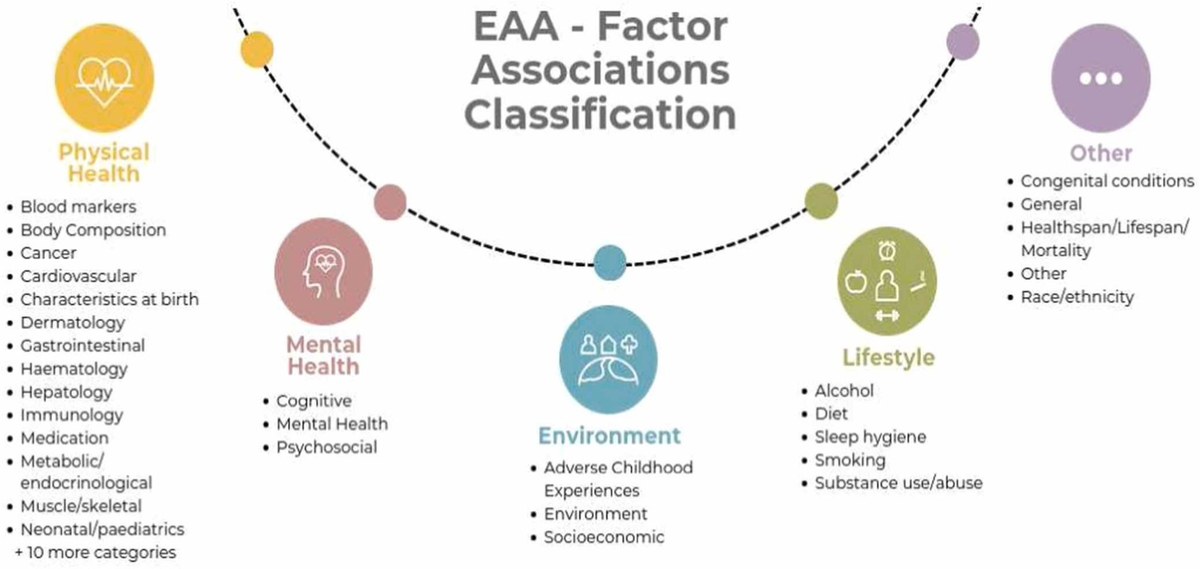
Figure 4. EAA associations (Chervova et al. (2024)).
DNA Methylation Profiling
Several technologies are available for DNA methylation profiling; however, Illumina Infinium microarrays remain the most widely used platform in population and epidemiological studies.
The current standard platform is the Illumina Infinium MethylationEPIC BeadChip Version 2 (EPIC v2), which interrogates approximately 1 million CpG sites across promoters, gene bodies, enhancers, CpG islands, shores, shelves, and other regulatory regions of the genome.
Following DNA extraction and bisulfite conversion, DNA methylation levels are quantified at individual CpG sites and imported into R as β-values (beta values). Beta values range from 0 to 1, where:
- 0 = fully unmethylated
- 1 = fully methylated
These beta values serve as the foundation for downstream analyses, including quality control, normalization, cell-type estimation, epigenetic clock calculation, and other DNA methylation-based biomarkers.
References & Resources
-
Aristizabal MJ, Anreiter I, Halldorsdottir T, et al. Biological embedding of experience across the lifespan: Epigenetic mechanisms and the missing heritability problem. Proceedings of the National Academy of Sciences. 2019;116(38):23970–23979.
-
Chervova O, van den Akker EB, Deelen J, et al. Ageing Research Reviews. 2024.
-
Teschendorff AE, Horvath S. Epigenetic ageing clocks: statistical methods and emerging computational challenges. Nature Reviews Genetics. 2025. https://www.nature.com/articles/s41576-024-00807-w
-
Merrill SM. Epigenetics and Psychosocial Interventions (EPI) Lab. https://drsarahmerrill.com/research/